Oxaziridine
| Oxaziridine | |

| |
| Identification | |
|---|---|
| Nom UICPA | Oxaziridine |
| No CAS | |
| PubChem | 15817734 |
| SMILES | |
| InChI | |
| Propriétés chimiques | |
| Formule | CH3NO [Isomères] |
| Masse molaire[1] | 45,040 6 ± 0,001 5 g/mol C 26,67 %, H 6,71 %, N 31,1 %, O 35,52 %, |
| Unités du SI et CNTP, sauf indication contraire. | |
| modifier |
|
Les oxaziridines sont une classe de composés organiques hétérocycliques à trois atomes, contenant un atome de carbone, un atome d'azote et un atome d'oxygène. L'oxaziridine CH2NHO est aussi le composé parent du groupe fonctionnel.

Structure générale des oxaziridines

Propriétés générales modifier
Les premiers dérivés d'oxaziridines sont synthétisés dans les années 1950 par Emmons[2], Krimm[3] et Horner et Jürgens[4]. Alors que les atomes d'oxygène et d'azote agissent usuellement comme nucléophiles du fait de leur électronégativité élevée, les oxaziridines permettent le transfert électrophile des deux hétéroatomes. Cette réactivité particulière est due à la présence du cycle à trois atomes très contraint, et de la relative faiblesse de la liaison N-O. Les nucléophiles ont tendance à attaquer sur l'azote de l'oxaziridine quand le substituant sur l'atome d'azote est petit (typiquement R1= H), et sur l'atome d'oxygène quand le substituant sur l'atome d'azote est très encombré stériquement. Ces effets électroniques peuvent être exploités pour réaliser différentes réactions de transfert d'atome d'oxygène ou d'azote, comme l'α-hydroxylation d'énolates, l'époxydation d'alcènes ou encore l'oxydation sélective de sulfures et de séléniures.
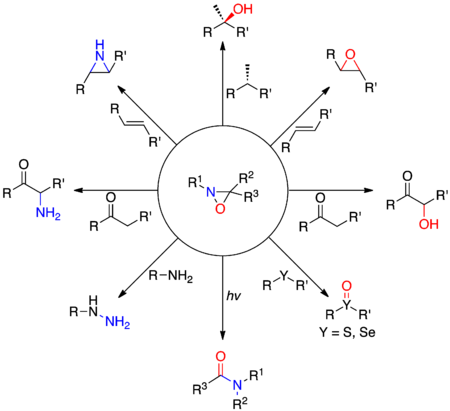
Schéma de quelques réactions utilisant les oxaziridines.

Chiralité modifier
Des réactifs chiraux de dérivés d'oxaziridines ont été développés, et permettent des réactions de transfert d'hétéroatomes stéréospécifiques. La chiralité des oxaziridines peut venir de la structure des substituants du cycle ou de la conformation de l'atome d'azote (invertomère). Les oxazirines sont en effet uniques du fait de la très haute barrière d'inversion de l'azote qui permet de conserver leur configuration stéréochimique. Cette barrière d'inversion à température ambiante est de l'ordre de 24 à 31 kcal·mol-1, et des oxaziridines énantiopures ont pu être préparées dans les années 1980[5].
Des dérivés d'oxaziridines à base de dérivés du camphre ont été synthétisés dans les années 1970[6], et sont devenus une pierre angulaire de la synthèse asymétrique. Parmi les nombreuses synthèses totales employant des oxaziridines, la synthèse totale du taxol par les équipes de Holton[7],[8] et de Wender[9],[10] proposent l'α-hydroxylation asymétrique avec le camphorsulfonyloxaxiridine comme une étape clé de la synthèse de cet agent anticancéreux.
Synthèses modifier
N-H, N-Alkyl, N-Aryloxaziridines modifier
Les deux principales stratégies de synthèse des N-H, N-Alkyl et N-Aryloxaziridines sont (A) l'oxydation d'imines par les peracides et (B) l'amination de carbonyles.

De plus, l'oxydation d'imines chirales et l'oxydation d'imines avec des peracides chiraux peut conduire à la préparation d'oxaziridines énantiomériquement pures.
N-Sulfonyloxaziridines modifier
Au début des années 1980, la première N-sulfonyloxaziridine est préparée[11]. Ces dérivés sulfonyles sont utilisés exclusivement pour les réactions de transfert d'atome d'oxygène, et sont actuellement la classe d'oxaziridines la plus communément employée. Originellement préparées à partir de m-CPBA et de chlorure de benzyltrimethylammonium comme catalyseur de transfert de phase, une amélioration de la synthèse permet désormais l'utilisation d'hydrogénopersulfate de potassium, ou Oxone comme oxydant[12].
De nombreux dérivés de N-sulfonyloxaziridines sont utilisés, chacun possédant différentes propriétés physico-chimiques et réactivités. Un résumé de ces différents réactifs est présenté dans le tableau suivant[13],[14],[15],[16],[17],[18],[19],[20],[21].

Oxaziridines perfluorées modifier
Les oxaziridines perfluorées présentent une forte réactivité comparées à leurs équivalents hydrocarbonés. Les substituants perfluorés sont électron-accepteurs, rendant la réactivité des oxaziridines perfluorées similaire à celle des dioxiranes[22]. Les perfluoroalkyloxaziridines peuvent notamment hydroxyler certaines liaisons C-H avec une très forte sélectivité. Les oxaziridines perfluorées peuvent être synthétisées en faisant réagir une imine perfluorée avec du peroxyde de perfluorométhylfluorocarbonyle et un métal fluoré pour piéger le HF libéré[22].

Réactions modifier
Transferts d'atome d'oxygène modifier
α-Hydroxylation des énolates modifier
Le groupement α-hydroxycétone, ou acyloïne, est un motif présent dans de nombreux produits naturels. De nombreuses voies de synthèse ont été employées pour reproduire ce motif, comme la réduction d'α-déicétones, la substitution d'hydroxyles pour un groupe partant, ou l'oxydation directe d'un énolate. Lors de cette dernière méthode, l'oxodiperoxymolybdenum(pyridine)-(triamide hexaméthylphosphorique) (MoOPH) et les N-sulfonyloxaziridines sont les sources d'oxygène électrophiles les plus communément employées. Les N-sulfonyloxaziridines présentent l'avantage d'induire une plus grande chiralité comparativement à l'utilisation de MoOPH et d'autres oxydants[23]. L'induction chirale a été démontrée avec de nombreuses autres cétones chirales et des cétones avec des auxiliaires de chiralité, comme le SAMP et le RAMP[13].
{kind=link}

Des travaux ont été menés sur l'hydroxylation asymétrique d'énolates prochiraux avec des dérivés de camphorsulfonyloxaziridine, permettant d'atteindre de bons excès énantiomériques[16]. Il est communément admis que l'état de transition qui permet d'obtenir cette bonne stéréochimie implique un état de transition ouvert dans lequel le groupement stériquement encombré détermine le côté par lequel la molécule est approchée[13].

La sélectivité de certaines hydroxylations peut être améliorée par l'ajout de groupements coordinants en position alpha du cycle oxaziridine, comme dans les oxaziridines 3b et 3c dans le tableau ci-dessus[19]. Il est proposé que la réaction passe par un état de transition fermé dans lequel l'oxyanion métallique est stabilisé par chélation grâce au sulfate et aux groupements coordinants du camphre[13].

L'α-hydroxylation avec des oxaziridines a été très largement utilisée en synthèse totale. C'est une étape clé dans la synthèse totale du Taxol par Holton (en) [24] et dans la synthèse totale du Taxol par Wender (en)[25]. De même, Forsyth utilise cette réaction dans la synthèse du système C3-C14 (1,7-dioxaspiro[5.5]undéc-3-ène substitué) de l'acide okadaïque[26]

Époxydation des alcènes modifier
L'époxydation des alcènes est très employée en synthèse organique, les époxydes pouvant être transformés en de nombreux groupes fonctionnels. Habituellement, l'époxydation utilise du m-CPBA (ou d'autres peracides), mais les oxaziridnes ont montré qu'ils peuvent être employés pour mener des synthèses similaires et sont très utiles pour préparer des époxydes sensibles aux conditions acidiques[5]. Ci-dessous est décrit la synthèse de la (-)-chaetominine qui utilise l'époxydation d'un alcène par une oxaziridine lors de l'une des dernières étapes de sa synthèse[27].

Une autre classe de transformation très employée en synthèse organique est l'époxydation asymétrique, comme l'époxydation de Sharpless, l'époxydation de Jacobsen ou l'époxydation de Juliá-Colonna (en). L'inconvénient majeur de ces réactions est qu'elles nécessitent des fonctionnalités très spécifiques pour permettre d'être sélectives. L'époxydation de Sharpless fonctionne avec des alcools allyliques, l'époxydation de Jacobsen avec des alcènes disubstitués en cis par des aromatiques, et l'époxydation de Juliá-Colonna des cétones α-β insaturées. L'utilisation d'oxaziridines asymétriques permet de réaliser des transformations stéréospécifiques sur des alcènes non-fonctionnalisés[5]. Le schéma ci-dessous présente l'époxydation asymétrique du trans-stilbène avec un sel d'oxaziridinium chiral utilisant de l'oxone comme oxydant[28].

Hydroxylation d'hydrocarbure désactivés modifier
Les oxaziridines perfluorées sont connues pour hydroxyler les hydrocarbures désactivés avec une grande régio- et diastéréospécificité[22]. Cette réaction est très importante, et peu d'autres composés présentent ce type de réactivité. Les oxaziridines perfluorées présentent une très bonne sélectivité vis-à-vis des carbones tertiaires. L'hydroxylation de carbones primaires ou la dihydroxylation de composés présentant deux sites oxydables n'a jamais été observée. La rétention de la stéréochimie est très importante, de l'ordre de 95 à 95 %, et peut être encore augmentée en ajoutant un sel de fluorure[29].

Transferts d'atome d'azote modifier
Moins d'attention a été donnée aux oxaziridines comme réactifs de transfert d'atome d'azote. Les oxaziridines non-substituées ou possédant un groupement acyle sur l'azote peuvent être utilisées pour de telles réactions, le premier exemple datant des années 1960[30].
Amination de N-nucléophiles modifier
L'amination de nucléophiles avec des oxaziridines non-substituées sur l'azote est assez polyvalent quant aux nucléophiles utilisables. Les hydrazines peuvent ainsi être préparées à partir d'amines secondaires ou tertiaires, les hydroxylamines et les thiohydroxylamines à partir d'alcools et de thiols, les sulfilimines à partir de thioéthers et les α-aminocétones à partir d'énolates[31].

N-acylamidation modifier
Le transfert d'amines acylées est plus difficiles que pour les amines non-substituées. Ces réactions de transfert ont tout d'abord été effectuées en utilisant des nucléophiles comme des amines et des hydrazines. Quelques rares exemples de transferts d'amines acylées vers des carbones nucléophiles ont aussi été décrits[31].

Réarrangements modifier
Les oxaziridines peuvent subir des réactions de réarrangement sous irradiation dans l'ultraviolet, via un mécanisme radicalaire, ou en présence d'un réducteur, comme le CuI. Les oxaziridines spirocycliques peuvent aussi étendre leur cycle pour donner la lactame correspondante[32]. La migration du substituant est contrôlée par des effets stéréoélectroniques, et le groupe en trans du doublet non liant de l'azote donne majoritairement le produit de migration[33]. Cet effet permet d'utiliser la chiralité de l'azote, du fait de la haute barrière d'inversion de l'azote, pour diriger le réarrangement, comme démontré sur le schéma ci-dessous. Sur la partie gauche du schéma le produit défavorable thermodynamiquement est observé de façon exclusive, alors que sur la partie droite du schéma le produit dérivé de l'intermédiaire radicalaire le moins stable est favorisé[32].

Ce type de réarrangement est utilisé dans une étape clé de la synthèse totale de la (+)-yohimbine[32], un médicament naturel pouvant être efficace contre l'impuissance sexuelle et les problèmes sexuels causé par les inhibiteurs sélectifs de la recapture de la sérotonine selon le National Institutes of Health[34].

Les oxaziridines peuvent aussi subir des réactions de réarrangement thermiques pour donner des nitrones. La sélectivité cis-trans est faible, mais les rendements sont bons. Il aussi est envisagé que certaines oxaziridines racémisent au cours du temps en passant par un intermédiaire nitrone[5].

Cycloaddions avec des hétérocumulènes modifier
Les oxaziridines peuvent réagir avec des hétérocumulènes pour donner différents hétérocycles à cinq atomes, comme décrit sur la figure ci-dessous. Cette réactivité est due à la contrainte du cycle à trois atomes et à la faiblesse de la liaison N-O[5].

Notes et références modifier
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- (en) W. D. Emmons, « The synthesis of oxaziranes », J. Am. Chem. Soc., vol. 78, no 23, , p. 6208–6209 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja01604a072).
- (de) H. Krimm, « Uber Isonitrone », Chem. Ber., vol. 91, no 5, , p. 1057-1068 (ISSN 0009-2940, DOI 10.1002/cber.19580910532).
- (de) L. Horner et E. Jürgens, « Notiz Über Darstellung und Eigenschaften Einiger Isonitrone (Oxazirane) », Chem. Ber., vol. 90, no 10, , p. 2184–2189 (ISSN 0009-2940, DOI 10.1002/cber.19570901010).
- (en) F. A. Davis et A. C. Sheppard, « Applications of oxaziridines in organic synthesis », Tetrahedron, vol. 45, no 18, , p. 5703–5742 (DOI 10.1016/S0040-4020(01)89102-X).
- (en) F. A. Davis, R. Jenkins et al., « 2-[(–)-Camphor-10-ylsulphonyl]-3-(nitrophenyl)oxaziridine: a new chiral oxidizing agent », J. Chem. Soc., Chem. Commun., , p. 600-601 (ISSN 0022-4936, DOI 10.1039/C39790000600).
- (en) R. A. Holton, C. Somoza et al., « First total synthesis of taxol. 1. Functionalization of the B ring », J. Am. Chem. Soc., vol. 116, no 4, , p. 1597–1598 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00083a066).
- (en) R. A. Holton, H. B. Kim et al., « First total synthesis of taxol. 2. Completion of the C and D rings », J. Am. Chem. Soc., vol. 116, no 4, , p. 1599–1600 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00083a067).
- (en) P. A. Wender, N. F. Badham et al., « The pinene path to taxanes. 5. Stereocontrolled synthesis of a versatile taxane Precursor », J. Am. Chem. Soc., vol. 119, no 11, , p. 2755–2756 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja9635387).
- (en) P. A. Wender, N. F. Badham et al., « The pinene path to taxanes. 6. A concise stereocontrolled synthesis of taxol », J. Am. Chem. Soc., vol. 119, no 11, , p. 2757–2758 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja963539z).
- (en) F. A. Davis et O. D. Stringer, « Chemistry of oxaziridines. 2. Improved synthesis of 2-sulfonyloxaziridines », J. Org. Chem., vol. 47, no 9, , p. 1774–1775 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo00348a039).
- (en) F. A. Davis, S. Chattopadhyay et al., « Chemistry of oxaziridines. 9. Synthesis of 2-sulfonyl- and 2-sulfamyloxaziridines using potassium peroxymonosulfate (oxone) », J. Org. Chem., vol. 53, no 9, , p. 2087–2089 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo00244a043).
- (en) F. A. Davis et B. C. Chen, « Asymmetric hydroxylation of enolates with N-sulfonyloxaziridines », Chem. Rev., vol. 92, no 5, , p. 919–934 (ISSN 0009-2665 et 1520-6890, DOI 10.1021/cr00013a008).
- (en) F. A. Davis et R. H. Jenkins, « Chemistry of oxaziridines. 3. Asymmetric oxidation of organosulfur compounds using chiral 2-sulfonyloxaziridines », J. Am. Chem. Soc., vol. 104, no 20, , p. 5412–5418 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00384a028).
- (en) F. A. Davis, R. T. Reddy et al., « Chemistry of oxaziridines. 15. Asymmetric oxidations using 3-substituted 1,2-benzisothiazole 1,1-dioxide oxides », J. Org. Chem., vol. 56, no 2, , p. 809–815 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo00002a056).
- (en) J. C. Towson, M. C. Weismiller et al., « (+)-(2R,8aS)-10-(Camphorylsulfonyl)oxaziridine », Org. Synth., vol. 69, , p. 158 (ISSN 0078-6209).
- (en) F. A. Davis et J. C. Towson, « Chemistry of oxaziridines. 11. (Camphorylsulfonyl)oxaziridine: synthesis and properties », J. Am. Chem. Soc., vol. 110, no 25, , p. 8477–8482 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00233a025).
- (en) R. D. Bach, B. A. Coddens et al., « The mechanism of oxygen transfer from an oxaziridine to a sulfide and a sulfoxide: a theoretical study », J. Org. Chem., vol. 55, no 10, , p. 3325–3330 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo00297a062).
- (en) F. A. Davis, A. Kumar et al., « Chemistry of oxaziridines. 16. A short, highly enantioselective synthesis of the AB-ring segments of γ-rhodomycionone and α-citromycinone using (+)-[(8,8-dimethoxycamphoryl)sulfonyl]oxaziridine », J. Org. Chem., vol. 53, no 3, , p. 1143–1145 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo00003a042).
- (en) F. A. Davis, M. C. Weismiller et al., « (Camphorylsulfonyl)imine dianion in the synthesis of new optically pure (camphorylsulfonyl)oxaziridine derivatives », Tetrahedron Lett., vol. 30, no 13, , p. 1613–1616 (ISSN 0040-4039, DOI 10.1016/S0040-4039(00)99534-0).
- (en) B. C. Chen, M. C. Weismiller et al., « Enantioselective synthesis of (+)-kjellmanianone », Tetrahedron, vol. 47, no 2, , p. 173–182 (ISSN 0040-4020, DOI 10.1016/S0040-4020(01)80914-5).
- (en) V. A. Petrov et G. Resnati, « Polyfluorinated oxaziridines: Synthesis and reactivity », Chem. Rev., vol. 96, no 5, , p. 1809–1824 (ISSN 0009-2665 et 1520-6890, DOI 10.1021/cr941146h).
- (en) D. A. Evans, M. M. Morrissey et al., « Asymmetric oxygenation of chiral imide enolates. A general approach to the synthesis of enantiomerically pure α-hydroxy carboxylic acid synthons », J. Am. Chem. Soc., vol. 107, no 14, , p. 4346–4348 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00300a054).
- (en) R. A. Holton, H. B. Kim et al., « First total synthesis of taxol. 2. Completion of the C and D rings », J. Am. Chem. Soc., vol. 116, no 4, , p. 1599–1600 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00083a067).
- (en) P. A. Wender, N. F. Badham et al., « The pinene path to taxanes. 5. Stereocontrolled synthesis of a versatile taxane precursor », J. Am. Chem. Soc., vol. 119, no 11, , p. 2755–2756 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja9635387).
- (en) A. B. Dounay et C. J. Forsyth, « Abbreviated synthesis of the C3−C14 (substituted 1,7-dioxaspiro[5.5]undec-3-ene) system of okadaic acid », Org. Lett., vol. 1, no 3, , p. 451–454 (ISSN 1523-7060 et 1523-7052, DOI 10.1021/ol9906615).
- (en) B. Malgesini, B. Forte et al., « A straightforward total synthesis of (−)-chaetominine », Chem. Eur. J., vol. 15, no 32, , p. 7922–7929 (ISSN 0947-6539 et 1521-3765, DOI 10.1002/chem.200900793).
- (en) L. Bohé, G. Hanquet et al., « The stereospecific synthesis of a new chiral oxaziridinium salt », Tetrahedron Lett., vol. 34, no 45, , p. 7271–7274 (ISSN 0040-4039, DOI 10.1016/S0040-4039(00)79306-3).
- (en) A. Arnone, S. Foletto et al., « Highly enantiospecific oxyfunctionalization of nonactivated hydrocarbon sites by perfluoro-cis-2-n-butyl-3-n-propyloxaziridine », Org. Lett., vol. 1, no 2, , p. 281–284 (ISSN 1523-7060 et 1523-7052, DOI 10.1021/ol990594e).
- (de) E. Schmitz, R. Ohme et al., « Isomere Oxime mit Dreiringstruktur », Chem. Ber., vol. 97, no 9, , p. 2521–2526 (ISSN 0009-2940, DOI 10.1002/cber.19640970916).
- (en) S. Andeae et E. Schmitz, « Electrophilic aminations with oxaziridines », Synthesis, no 5, , p. 327-341 (ISSN 0039-7881 et 1437-210X, DOI 10.1055/s-1991-26459).
- (en) J. Aubé, « Oxiziridine rearrangements in asymmetric synthesis », Chem. Soc. Rev., vol. 26, no 4, , p. 269-277 (ISSN 0306-0012 et 1460-4744, DOI 10.1039/CS9972600269).
- (en) A. Lattes, E. Oliveros et al., « Photochemical and thermal rearrangement of oxaziridines. Experimental evidence in support of the stereoelectronic control theory », J. Am. Chem. Soc., vol. 104, no 14, , p. 3929–3934 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00378a024).
- « Yohimbe », sur le site nlm.nih.gov (consulté le ).
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Oxaziridine » (voir la liste des auteurs).