Leucémie aigüe myéloïde
Ne doit pas être confondu avec Leucémie myéloïde chronique.

| Causes | radiations, benzène |
|---|---|
| Symptômes | infections, saignements |
| Complications | réaction du greffon contre l'hôte (GvH) |
| Diagnostic | hémogramme, myélogramme, cytogénétique |
|---|---|
| Traitement | chimiothérapie anticancéreuse, thérapie ciblée, allogreffe de moelle osseuse |
| Médicament | Idarubicine, mitoxantrone, étoposide, (RS)-cyclophosphamide, daunorubicine, hydrate de thioguanine (d), gemtuzumab ozogamicin, dasatinib monohydraté (d), énasidénib, vénétoclax, doxorubicine, hydroxyurée, cytarabine, évérolimus, azacitidine, méthotrexate, déférasirox, trétinoïne, nivolumab, clofarabine, décitabine, midostaurine, busulfan, sorafénib, ruxolitinib, 6-thioguanine et ponatinib |
| Spécialité | Oncologie et hématologie |
| CIM-10 | C92.0 |
|---|---|
| CIM-9 | 205.0 |
| ICD-O | M9861/3 |
| OMIM | 601626 |
| DiseasesDB | 203 |
| MedlinePlus | 000542 |
| eMedicine | 197802 |
| MeSH | D015470 |
| Patient UK | Acute-myeloid-leukaemia-pro |
La leucémie aigüe myéloïde (LAM), aussi appelée leucémie aigüe myéloblastique, est un cancer de type hémopathie maligne affectant les cellules hématopoïétiques de la moelle osseuse. Les cellules leucémiques, appelées blastes, sont caractérisées par une incapacité à se différencier en cellules matures et par une prolifération incontrôlée[1]. Ce dysfonctionnement de la moelle osseuse empêche la production normale des cellules sanguines et se traduit par divers syndromes cliniques, parfois très graves (infection, hémorragie, etc.). Les LAM surviennent classiquement chez l'adulte au-delà de 40 ans, en particulier chez le sujet âgé même si elles sont également recensées parmi les cancers pédiatriques. La prise en charge est assurée par des médecins hématologues, majoritairement en milieu hospitalier. Le diagnostic fait appel à la biologie moléculaire pour identifier notamment des mutations génétiques, ce qui permet de déterminer le sous-type de LAM. Ces éléments diagnostics entrent ensuite en ligne de compte pour établir le pronostic de la maladie avec d'autres éléments comme l'âge ou la réponse au traitement. L'association de la chimiothérapie intensive et de l'allogreffe de moelle osseuse permet d'obtenir des rémissions complètes et durables voire des guérisons chez environ 20 à 40 % des patients[2],[3].
Domaine d'intérêt pour la recherche médicale, des progrès importants ont été accomplis au cours des années 2000 et 2010. L’avènement de nouvelles techniques performantes en hématologie cellulaire, en biologie moléculaire et en cytogénétique a donné accès à une meilleure compréhension des mécanismes impliqués dans le développement des LAM. L'identification de nouvelles cibles thérapeutiques permet de proposer des thérapies ciblées en alternative à la chimiothérapie cytotoxique. Ainsi, pour un sous-type particulier, la leucémie aigüe promyélocytaire, la chimiothérapie classique tend à ne plus être proposée en première ligne de traitement.
Épidémiologie modifier
Incidence modifier
Les LAM peuvent survenir à tout âge mais affectent surtout l'adulte après 50 ans. Durant la décennie 2010, l'âge médian de diagnostic était compris entre 64 et 72 ans selon les pays et les années.
| Épidémiologie des LAM | ||||
| Paramètre | France[4] | Europe[5],[6] | États-Unis[7] | |
|---|---|---|---|---|
| Prévalence (pour 100 000 hab.) | 11[8] | |||
| Incidence (pour 100 000 hab.) | 2,3 (F) / 3,1 (H) (2018) | 2 - 3 | 3,5 (F) - 5,2 (H) (2012-2016) | |
| Sex ratio (H/F) | 1,3 | 1,2 | 1,5 | |
| Proportion des hémopathies | 7,6 % | |||
| Proportion des cancers | 0,9 % | 1,2 % | ||
La population pédiatrique (0-19 ans) est peu affectée par les LAM : l'incidence est faible avec, en 2017, une incidence de 0,9 %[9]. Ces patients sont davantage touchés par un autre sous-type de leucémie : la leucémie aiguë lymphoblastique qui représente environ 80 % des cas de leucémie de l'enfant contre 20 % pour les LAM[10].
Mortalité modifier
Le taux de mortalité standardisé sur l'âge lié aux LAM est estimé à 1,3 pour 100 000 personnes en 2017 par le Global Burden of Disease[11]. Pour la population pédiatrique (0 à 19 ans) il est plus faible à 0,4 pour 100 000 personnes.
Pathogénie, causes, facteurs de risque modifier
Pathogénie modifier
Les hémopathies malignes, dont les LAM font partie, peuvent être globalement caractérisées par deux attributs : la maturation et la prolifération. Dans les LAM, ces deux caractéristiques sont altérées : les blastes ont perdu leur capacité de différentiation en cellules sanguines matures (polynucléaires, monocytes, plaquettes, globule rouge) et la division cellulaire s'effectue sans contrôle avec une prolifération excessive. Cette maturation et prolifération défaillantes sont liées à un défaut dans le cycle cellulaire : généralement certains gènes chargés de contrôler ces fonctions ont subi des mutations qui perturbent le contrôle physiologique de la division des cellules. L'identification de ces altérations servent de base au diagnostic, à la classification et au traitement des LAM.

Causes modifier
Les cancers et les leucémies n'ont pas de cause unique connue, mais leur développement est cependant associé à certains facteurs environnementaux et/ou anomalies génétiques. Le Centre international de recherche sur le cancer, une agence de l'OMS, est notamment chargé de l'évaluation et classification des agents physiques, chimiques et biologiques en fonction de leur effet cancérogène. Certains d'entre eux favorisent particulièrement l'apparition de leucémies dont les LAM[12],[13]. Globalement, toute exposition pouvant altérer, modifier ou dénaturer l'ADN des cellules est un facteur de risque. Cependant, des données épidémiologiques et des études des effets cellulaires sont nécessaires pour évaluer, avec un degré raisonnable de confiance, le risque présenté par une telle exposition.
Rayonnements ionisants modifier
Les rayonnements ionisants sont des particules assez énergétiques pour casser les liaisons chimiques entre atomes et donc altérer les molécules. La découverte de la radioactivité et son utilisation croissante au cours du XXe siècle ont fait rapidement apparaître les risques de l'exposition aux radiations pour la santé. Le développement de la radiobiologie a par la suite permis d'établir que radiations provoquent des anomalies au niveau de l'ADN, et augmentent le risque de survenue de cancer et particulièrement de leucémies. Marie Curie et Irène Joliot-Curie sont ainsi décédées de leucémies probablement radio-induites du fait de leur travaux sur la radioactivité[14],[15].
L'étude INWORKS, publiée dans le Lancet Haematology en 2015, portant sur une cohorte d'un peu plus de 300 000 travailleurs du nucléaire dans 3 pays, conclut à une augmentation du risque de leucémie même lors de faibles expositions[16].
Agents chimiques modifier
Des agents chimiques dont des médicaments et en particulier les chimiothérapies cytotoxiques sont identifiés comme pourvoyeurs de leucémies.
L'exposition au benzène est une cause connue de développement de LAM ; il est ainsi classé depuis 1979 comme cancérogène du groupe 1 par le CIRC[17],[18],[19] (cancérogène avéré).
Le glyphosate est classé comme probablement cancérogène pour l’humain (groupe 2A) mais il n'est pas retrouvé de lien avec les LAM avec un fort niveau de preuve[20] ; bien que les données soient limitées, un lien reste suspecté[21],[22].
Médicaments identifiés comme leucémogènes[23] :
- les anthracyclines (doxorubicine, daunorubicine, etc.) et apparentés (mitoxantrone) sont des molécules anticancéreuses connues pour être leucémogènes ;
- les agents alkylants (busulfan, etc.) ;
- les antimétabolites (cytarabine) ;
- les épidophylotoxines : étoposide[24].
- Agent chimiques leucémogènes
-
 Benzène
Benzène -
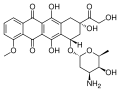 Doxorubicine
Doxorubicine -
 Étoposide
Étoposide



Causes biologiques modifier
L'apparition d'une leucémie aigüe myéloïde peut faire suite à la transformation (ou acutisation[a]) d'une autre hémopathie comme un syndrome myélodysplasique ou un syndrome myéloprolifératif (leucémie myéloïde chronique, etc.). Diverses maladies congénitales constituent un facteur de risque (trisomie 21, syndrome de Klinefelter, anémie de Fanconi, etc.)[25],[26].
Les antécédents d'infection par certains virus ou bactéries ne semblent pas reliés à un risque accru de LAM.
Signes cliniques modifier
Lorsque les blastes prolifèrent excessivement ils envahissent la moelle osseuse et/ou d'autres organes du corps humain et perturbent leur fonctionnement, causant l'apparition des signes cliniques. Cependant, la présentation clinique est généralement peu spécifique et le diagnostic ne peut être posé sur les seuls symptômes [27],[28],[29],[30].
Insuffisance médullaire modifier
L'envahissement de la moelle osseuse par les cellules leucémiques va perturber l'hématopoïèse c'est-à-dire le processus physiologique de création de nouvelles cellules sanguines. Trois types de cellules (ou lignées) sont habituellement produites dans la moelle : les globules rouges (érythrocytes), les globules blancs (notamment les polynucléaires neutrophiles) et les plaquettes (thrombocytes). Le dysfonctionnement de la moelle peut aller de la cytopénie (une seule lignée cellulaire touchée) à l'aplasie médullaire (déficit complet en cellules sanguines des trois lignées). Les symptômes associés dépendent du type de cytopénie. Les cytopénies peuvent être diagnostiquées devant la manifestation de symptômes cliniques ou bien sur un bilan biologique de routine chez un individu asymptomatique.
Atteinte de la lignée rouge modifier
Un déficit quantitatif en globules rouges est objectivé par la concentration totale d'hémoglobine dans le sang et est appelé anémie. La tolérance du patient à l'anémie est généralement liée à la vitesse d'apparition : en cas d'installation à bas bruit, l'organisme s'adapte via des systèmes de compensation les symptômes peuvent être absents. Un déclenchement rapide s'accompagnera au contraire d'un retentissement clinique avec notamment dyspnée, asthénie, pâleur.
Atteinte de la lignée blanche modifier
La diminution des polynucléaires neutrophiles (PNN) pose le principal problème : ces cellules appartiennent au système immunitaire inné et forment la première ligne de défense contre les infections, principalement bactériennes et fongiques. Selon l'importance du déficit on parlera de neutropénie ou d'agranulocytose. Le sujet neutropénique est extrêmement vulnérables aux infections. L'apparition d'une fièvre dans ce contexte est appelée neutropénie fébrile, et constitue une urgence thérapeutique nécessitant le plus souvent une prise en charge en milieu hospitalier avec administration d'un antibiotique.
On peut parler de leucémie « hyperleucocytaire » (c'est-à-dire avec excès de globules blancs) tout en observant une neutropénie : un pourcentage élevé de leucocytes correspond en réalité aux blastes circulant dans le sang avec, parallèlement, une chute du nombre polynucléaires normaux.
Atteinte de la lignée plaquettaire modifier

Le diminution du nombre de plaquettes dans le sang expose à un risque de saignements importants ou spontanés. Cliniquement, des signes cutanés comme un purpura, des pétéchies ou des ecchymoses peuvent traduire l'inefficacité de l'hémostase.
Des troubles de la coagulation importants sont en particulier observés lors du stade initial de la leucémie aigüe promyélocytaire. Une consommation des plaquettes et des facteurs de coagulation entraînée par la coagulation intravasculaire disséminée peut aggraver la thrombopénie.
Syndrome tumoral modifier
Le syndrome tumoral est lié à la dissémination des blastes hors de la moelle osseuses et de la accumulation dans les organes. Il peut se traduire cliniquement par[31] :

.jpg)
- une augmentation de volume de certains organes : splénomégalie, hépatomégalie ;
- des douleurs articulaires ;
- une atteinte cutanée (sudation nocturne) ;
- une atteinte muqueuse avec notamment une hypertrophie gingivale (augmentation du volume des gencives) ;
- une atteinte neuroméningée lors de l'envahissement du système nerveux central, pouvant être accompagnée de signes neurologiques.
Lorsque la masse tumorale (c'est-à-dire le nombre de blastes) est très importante, un syndrome de lyse tumorale peut se déclencher. Il est lié à la destruction des blastes qu'elle soit spontanée ou liée à un traitement. Le contenu intracellulaire est alors relargué dans le sang ce qui entraîne des perturbations des concentrations sanguines en ions (potassium, phosphate), visibles sur le ionogramme. Cela peut entraîner une hyperkaliémie, délétère pour le rythme cardiaque ou une élévation de l'acide urique (hyperuricémie) pouvant conduire à des atteintes rénales via la formation de cristaux d'urate. La prise en charge du syndrome de lyse vise à rétablir un ionogramme normal par hydratation et administration d'électrolytes ; l'hyperuricémie peut être traitée par rasburicase, une enzyme qui dégrade l'acide urique.
Le syndrome de leucostase est observé en cas d'infiltration massive des organes par les cellules leucémiques. Il se manifeste par une forte concentration des blastes dans le sang (blastose), une fièvre et des symptômes respiratoires et ou neurologiques. Ce syndrome est une urgence thérapeutique dont l'issue peut être fatale par détresse respiratoire.
Diagnostic modifier
L'examen clinique du patient permet surtout de dépister des signes d'hémopathie maligne. Le diagnostic repose essentiellement sur un bilan biologique élargi comprenant : hémogramme (NFS), myélogramme ou biopsie ostéomédullaire, immunophénotypage, caryotype, cytogénétique et séquençage de gènes[32].

L'examen de la moelle osseuse permet de définir l'aspect morphologique des cellules et est la première étape d'orientation du diagnostic. Le diagnostic de leucémie aigüe est posé en présence de plus de 20 % de cellules immatures, les blastes. L'examen de l'expression de divers récepteurs (les CD) par immunophénotypage permet de classifier le stade et la lignée à laquelle les cellules appartiennent.

Le caryotype et l'étude du génome permettent l'identification d'anomalies chromosomiques (délétions, inversions, translocations) ; 50 à 60 % des LAM présentent des anomalies du caryotype. Il est également recherché des mutations génétiques. Ces analyses sont déterminantes pour évaluer le pronostic et les options thérapeutiques.
Avant le développement des techniques de phénotypage et de cytogénétique, le diagnostic des LAM s'appuyait majoritairement sur la morphologie cellulaire et était guidé par la classification French-American-British (FAB). Les nouvelles techniques d'étude de l'ADN ont permis de constituer la nouvelle classification OMS qui a remplacé la FAB : sa catégorisation est plus fine et s'appuie sur les anomalies chromosomiques et/ou génétiques.
En dehors des classifications, on peut aussi distinguer 3 types de LAM[33] :
- les LAM de novo ;
- les LAM secondaires à un syndrome myélodysplasique ou à un syndrome myéloprolifératif ;
- les LAM induites par les cytotoxiques et/ou les radiations.
Classification modifier
De par les cellules affectées et les mutations en jeu, les leucémies aigües myéloïdes sont un ensemble disparate qui a fait l'objet de catégorisation. La FAB (French-American-British) est la plus ancienne et a été remplacée par la classification OMS parue en 2008. Bien que la FAB soit obsolète, il est encore habituel de rencontrer les termes diagnostics s'y rapportant (LAM0, LAM3, etc.).
Organisation Mondiale de la Santé modifier
La classification OMS des LAM est celle actuellement utilisée pour les diagnostics. Initialement publiée en 2008, elle a été actualisée en 2016 et publiée dans Blood (revue d'hématologie de référence)[34].
| Classification OMS 2016 des leucémies aigües myéloïdes et néoplasies liées | ||
| Nom | Description | ICD-O |
|---|---|---|
| LAM avec anomalies cytogénétiques récurrentes |
|
Multiple |
| LAM avec anomalies associées aux myélodysplasies | Regroupe les LAM associées aux SMD dépourvues des anomalies moléculaires précédemennt citées ; elles sont généralement associées à un pronostic défavorable.
|
M9895/3 |
| LAM post-traitement | Groupe incluant les leucémies survenant après chimiothérapie ou exposition aux radiations. | M9920/3 |
| LAM, sans autre indication | Groupe incluant les sous-types n'entrant pas dans d'autres catégories. | |
| Sarcome myéloïde | ||
| Proliférations myéloïdes associées à la trisomie 21 |
|
M9861/3 |
| Leucémie dérivée des cellules dendritiques plasmocytoïdes | Autrefois connu sous le nom leucémie/lymphome à cellules NK
Également appelée : néoplasme à cellules dendritiques plasmacytoïdes blastiques (NCDPB) |
|
| Leucémies aigües de lignée ambigüe |
|
|
FAB (French-American-British) modifier
La classification FAB (French-American-British, 1976)[35] est l'ancienne classification, aujourd'hui abandonnée. Elle se base sur la morphologie et la quantité de cellules observées sur le myélogramme.
| Classification FAB des LAM | ||
| LAM | Description | Proportion |
|---|---|---|
| LAM 0 | indifférenciée | 5 % des cas |
| LAM 1 | myéloblastique sans différenciation | 15 % des cas |
| LAM 2 | myéloblastique avec différenciation | 25 % des cas |
| LAM 3 | promyélocytaire | 10 % des cas |
| LAM 4 | myélomonocytaire | 20 % des cas |
| LAM 4Eo | myélomonocytaire avec éosinophilie | 5 % des cas |
| LAM 5 | monoblastique (sans différenciation : M5a, avec différenciation : M5b) | 10 % des cas |
| LAM 6 | érythroblastique ou érythroleucémie | 5 % des cas |
| LAM 7 | mégacaryoblastique | 5 % des cas |
Cette classification n'a pas d'impact en termes de pronostic ou de traitement à mettre en œuvre à la différence d'une analyse portant sur l'étude des gènes mutés[36].
Traitement modifier
Le traitement curatif des leucémies aigües myéloïdes est la chimiothérapie intensive associée à l'allogreffe de cellules souches hématopoïétiques[37] (à l'exception de la leucémie aigüe (« LA ») promyélocytaire). La prise en charge dépend de nombreux éléments liés à la fois au patient et à la leucémie. L'âge, l'état général, les comorbidités et les souhaits du patient sont notamment des facteurs prépondérants. Le type de leucémie aigüe, les différentes mutations détectées ainsi que les réponses cliniques aux traitements antérieurs déterminent les options de traitement tout au long de la prise en charge. Les décisions thérapeutiques sont prises à l'issues de réunions de concertation pluridisciplinaire (RCP)[38],[39].
Le traitement classique est une succession de cures de chimiothérapie en plusieurs phases : l'induction, la consolidation puis, si le patient est éligible, l'intensification thérapeutique avec allogreffe de moelle osseuse. La radiothérapie n'est pas un traitement standard des LAM étant donné qu'il n'existe pas de localisation unique de la maladie comme pour une tumeur solide. Elle est cependant utilisée pour prévenir ou traiter une atteinte neuro-méningée, c'est-à-dire lorsque les cellules leucémiques sont retrouvées dans le système nerveux central (cerveau, moelle épinière)[40]. D'autres situations particulières peuvent nécessiter un recours à la radiothérapie (localisation testiculaire, sarcome myéloïde)[41].
Le traitement de la leucémie aigüe promyélocytaire (LAP) est un cas particulier : le rôle de la chimiothérapie cytotoxique classique est moins prépondérant depuis le développement de l'association de l'acide tout-trans rétinoïque (ATRA) et du trioxyde d'arsenic permettant d'atteindre des taux de rémissions supérieurs à 90 %[42].
La prise en charge des leucémies et du cancer est associée à de nombreux soins de supports indispensables[43],[44] : prévention et gestion des effets indésirables des chimiothérapies, support transfusionnel pour pallier les cytopénies, suivi nutritionnel, dépistage de symptômes dépressifs, traitement de la douleur et mise en place des soins palliatifs adaptés si nécessaires.
Induction modifier
L'induction vise à induire une rémission complète (CR) sur les plans clinique et biologique par la chimiothérapie. Le protocole 3+7, à base de cytarabine et d'anthracycline (daunorubicine ou idarubicine), est classiquement prescrit[45] ; ce traitement provoque chez le patient une aplasie médullaire profonde de trois à quatre semaines, durant lesquelles ce dernier doit bénéficier d'une surveillance rapprochée dans un service d'hématologie spécialisé afin de prévenir les risques hémorragiques et infectieux.
En alternative à l'induction classique, d'autres molécules peuvent être employées comme la décitabine[46].
Consolidation modifier
La suite du traitement dépend dans une large mesure du pronostic de la LAM, de la réponse à la chimiothérapie d'induction (obtention d'une rémission complète), du patient (âge, comorbidités, etc.) et de ses souhaits de traitement. Les traitements de consolidation visent à éliminer les cellules leucémique résiduelles et à ainsi prévenir les récidives[47]. L'allogreffe de moelle osseuse est le traitement préférentiel permettant d'atteindre le taux de rémissions durables le plus élevé mais au prix d'une prise en charge assez lourde.
Si l'allogreffe est refusée ou inappropriée, la chimiothérapie standard peut être poursuivie, par exemple par cytarabine haute dose, azacitidine ou clofarabine. Les autres options thérapeutiques sont les thérapies ciblées. L'autogreffe de moelle osseuse est très rare[48].
Allogreffe modifier
L'allogreffe de moelle osseuse est le seul traitement curatif des LAM. L'objectif est de détruire la moelle osseuse pathologique du patient puis de reconstituer une nouvelle moelle grâce à des cellules souches hématopoïétiques collectées chez un individu sain. La prise en charge thérapeutique est complexe et requiert un service d'hématologie adapté disposant de chambres protégées ainsi que d'autres infrastructures hospitalières (unité de thérapie cellulaire, unité de préparation des chimiothérapies, etc.)[49].
L'éligibilité à la greffe est évaluée collégialement et décidée en accord avec le patient. Les traitements nécessaires à la greffe possèdent une certaine toxicité et induisent une longue période d'aplasie et d'immunodépression avec un risque de complications et d'infections élevé[50],[51]. L'allogreffe est donc généralement réservée aux sujets « jeunes » (moins de 60 ans) et/ou en très bon état général qui pourront supporter cette prise en charge et tirer un bénéfice à long terme de la greffe[52].
On peut distinguer sommairement 3 étapes[53]. Premièrement, le conditionnement du patient consiste en une irradiation corporelle totale[54], l'administration d'immunosuppresseurs et une chimiothérapie intensive. Il vise à éradiquer les cellules leucémiques ainsi que l'induction d'une immunodépression importante permettant de diminuer au maximum un rejet de greffe. Les cellules souches hétérologues sont ensuite réinjectées. La suite de la prise en charge consiste à gérer l'aplasie médullaire prolongée induite par le conditionnement ainsi que les éventuelles complications et des effets indésirables. À moyen terme, l'hématologue réalise un suivi de la santé du greffon et surveille la disparition effective des cellules leucémiques.
Au niveau immunologique, l'introduction dans l'organisme de cellules étrangères au patient (dites du « non-soi ») peut déclencher une réaction immunitaire dirigée contre les cellules du patient ou la leucémie : ces effets sont respectivement nommés « greffon vs. hôte » (GvH) et « greffon vs. leucémie » (GvL). Le premier est un effet indésirable plus ou moins grave qui peut se manifester en diverses localisations (digestive, cutanée, etc.) et correspond à une attaque des organes de l'hôte par les cellules greffées ; elle peut être aigüe ou chronique. L'effet GvL est au contraire bénéfique puisque les cellules immunitaires du greffon vont attaquer les cellules leucémiques et exercer ainsi un effet anticancéreux[55]. Outre la GvH, les complications de l'allogreffe incluent : infections en lien avec l'immunodépression et l'aplasie, augmentation du risque de cancers secondaires, cytopénies durables nécessitant des transfusions[53], maladie veino-occlusive hépatique ainsi que les autres effets indésirables des chimiothérapies.
Pour les patients fragiles ou âgés ne pouvant supporter une allogreffe standard, il existe des protocoles dits « d'intensité réduite » ou l'étape de conditionnement (chimiothérapie et radiothérapie) comporte des doses diminués : cette solution est un compromis pour préserver le bénéfice de l'allogreffe tout en prenant en compte la moindre tolérance tolérance du patient [56].
En 2016, en France, un peu plus de 700 allogreffes ont été réalisées pour traiter des LAM[48]. En 201, la société européenne pour la transplantation de sang et de moelle (EBMT) a recensé 6943 allogreffes et 293 autogreffes ayant une leucémie aiguë myéloïde pour indication[57].
Thérapies ciblées modifier
Lors du diagnostic, le séquençage du génome des cellules leucémique permet d'identifier les éventuelles mutations présentes. Certaines d'entre elles participent à la prolifération et au défaut de maturation des blastes : elles peuvent, par exemple, mener à la synthèse d'une protéine anormale qui activerait en permanence le cycle cellulaire, entraînant ainsi une division incontrôlée des cellules. L'enrayement de l'action néfaste des gènes mutés ou des protéines altérées constitue donc une cible thérapeutique potentielle : c'est ainsi que certains médicaments agissant spécifiquement sur ces anomalies ont été développés en cancérologie. Ils sont regroupés sous l'appellation thérapies ciblées en opposition à la chimiothérapie cytotoxique classique ; cette désignation inclut également d'autres médicaments au mécanisme d'action différent, par exemple les anticorps monoclonaux ciblant les récepteurs à la surface des cellules (les cluster de différentiation).
En 2016, une revue de la littérature estime que 50 % à 80 % des cas de LAM possèderaient des mutations délétères[58]. Parmi les gènes le plus souvent concernés on peut citer FLT3 ou NPM1[59] (les mutations peuvent être concomitantes). Dans le cadre de la prise charge d'un patient, la connaissance des gènes mutés permet non seulement de catégoriser la leucémie, d'établir un pronostic mais également de prévoir quelles thérapies ciblées pourront être prescrite chez ce malade. Bien qu'il n'existe pas encore une molécule pour contrer chaque gène, quelques-unes ont été développées au cours de la décennie 2010 et ont parfois reçu l'approbation des autorités sanitaires[60]. Par exemple, il a été identifié que des mutations de l'isocitrate déshydrogénase (IDH), une enzyme impliquée dans le métabolisme énergétique cellulaire, peuvent promouvoir la prolifération des cellules leucémiques. Ainsi, il a été développé des inhibiteurs de l'IDH : l'énasidénib (ciblant l'IDH2) et l'ivosidénib (ciblant l'IDH1)[61],[62]. Diverses autres thérapies ciblées peuvent être prescrites ou font l'objet de recherches cliniques comme le glasdegib, le venetoclax, etc.
| Cible | Type | Exemple de thérapie ciblée |
|---|---|---|
| FLT3 | Protéine | Midostaurine[63], giltéritinib (en)[64], quizartinib (en) |
| IDH | Protéine | Ivosidénib (IDH1), énasidénib (IDH2) |
| Bcl-2 | Protéine | Vénétoclax |
| CD33 | Récepteur membranaire | Gemtuzumab ozogamicine |
L'utilisation d'hormones androgèniques – qui ne sont pas à proprement parler des thérapies ciblées mais relève plutôt de l'hormonothérapie – a été également étudiée en recherche clinique. La noréthandrolone semble notamment améliorer la survie et la rémission des patients âgés[65],[39].
Enfin, certaines chimiothérapies, bien qu'ayant un mécanisme classique interférent avec la synthèse de l'ADN peuvent aussi avoir des propriétés additionnelles ciblées. L'azacitidine possède par exemple une action hypométhylante : elle peut induire un retrait de groupement chimiques méthyles (-CH3) des brins d'ADN et permettre ainsi la ré-expression de certains gènes contrôlant la division cellulaire de la cellule[66].
Autres situations modifier
Les formes récidivantes ou réfractaires des LAM sont traitées par des protocoles spécifiques avec généralement de la chimiothérapie (traitement de rattrapage ou de sauvetage)[38],[39].
Concernant l'immunothérapie, le gemtuzumab ozogamicine, est le seul anticorps monoclonal approuvé dans le traitement des LAM[67]. Les inhibiteurs d'immunocheckpoint (comme l'ipilimumab, le nivolumab) ou les cellules T CAR (lymphocytes modifiés par génie génétique) ne sont pas, en 2020, indiquées dans le traitement des LAM mais sont un sujet d'étude pour la recherche clinique[68],[69],[38].
Survie modifier
Le pronostic est généralement exprimé par la survie globale à 5 ans, c'est-à-dire par la proportion de patients toujours en vie 5 années après leur diagnostic, en prenant en compte toutes les causes de décès confondues (qu'elles soient liées à la LAM, ses conséquences, à un autre évènement sans lien comme par ex. un accident de la route). En oncologie, d'autres moyens peuvent être employés pour estimer le pronostic lors de la prise en charge ou dans les essais cliniques : on peut parler en termes de survie sans rechute, survie sans progression ou bien de la réponse au traitement (réponse complète, partielle, etc.).
Chez l'adulte, la survie à 5 ans est estimée à environ 25%[70],[71] et décroit avec l'âge : elle est de 40% avant 65 ans puis de 5% après[72]. En pédiatrie, la probabilité de survie globale à 5 ans a progressé de 50 % dans les années 90 à environ 60-70 % en 2010 (dans les leucémies aigües lymphoblastiques, elle atteint 90 % en 2010)[73],[74]. Là aussi, les chances de survie des jeunes enfants sont légèrement supérieures à celles des adolescents[72].
Ces chiffres ne sont pas applicables pour la leucémie aigüe promyélocytaire (ex-LAM 3). Auparavant associée à un pronostic sombre, de nouveaux traitements (association ATRA / ATO) permettent depuis les années 2000 d'obtenir des rémissions à long-terme chez 90% des patients[75].
Histoire modifier

La première description dans la littérature médicale d'un cas de leucémie remonte à 1827. Alfred Velpeau décrivit le cas d'un fleuriste âgé de 63 ans atteint d'une maladie et présentant fièvre, fatigue, des calculs rénaux et une importante dilatation du foie et de la rate. Il observa que le sang du patient avait une consistance de gruau et postula que cette apparence pouvait être causé par des particules blanches[76]. En 1845, une série de cas de patients décédés avec des rates dilatées et des « changements de couleur de consistance de leur sang » fut rapporté par le pathologiste J.H. Bennett ; il emploie alors le terme « leucocythémie » pour décrire cette affection[77].
Le mot « leucémie » fut employé pour la première fois par Rudolf Virchow, médecin pathologiste allemand, en 1856. Il décrivit, grâce à un microscope, un excès de globules blancs chez des individus présentant les symptômes décrits par Velpeau et Benett. Virchow ne connaissant pas l'origine de cette anomalie, il la nomma par un terme purement descriptif leucémie (en grec : sang blanc)[78].
Notes et références modifier
Notes modifier
- Anglicisme provenant de acute, « aigu », signifiant le passage d'une maladie chronique à un stade aigu ; le verbe associé est « acutiser » (aiguiser).
Références modifier
- Grand Dictionnaire Terminologique, Office québécois de la langue française, « leucémie aiguë », (consulté le )
- MSD, Ashkan Emadi, Jennie York Law, « Leucémie myéloïde aiguë (LMA) - Hématologie et oncologie - Édition professionnelle du Manuel MSD », (consulté le )
- (en) National Cancer Institute, « Adult Acute Myeloid Leukemia Treatment (PDQ®)–Health Professional Version », (consulté le )
- Institut National Du Cancer, « Synthèse - Estimations nationales de l'incidence et de la mortalité par cancer en France métropolitaine entre 1990 et 2018 - Ref : SYNINCNAT2019 », sur www.e-cancer.fr, (consulté le )
- INSERM, « Orphanet: Leucémie aiguë myéloïde », sur orpha.net, (consulté le )
- (en) D. Weber, E. Fromm, S. Erhardt, M. Lübbert, W. Fiedler, T. Kindler, J. Krauter, P. Brossart, A. Kündgen, H. R. Salih, J. Westermann, G. Wulf, B. Hertenstein, M. Wattad, K. Götze, D. Kraemer, T. Heinicke, M. Girschikofsky, H.G. Derigs, H. A. Horst, C. Rudolph, M. Heuser, G. Göhring, V. Teleanu, L. Bullinger, F. Thol, V. I. Gaidzik, P. Paschka, K. Döhner, A. Ganser, Hartmut Döhner, R. F. Schlenk, German-Austrian AML Study Group (AMLSG) Gabriele Nagel, « Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO) », Annals of Hematology, vol. 96, no 12, , p. 1993 (DOI 10.1007/s00277-017-3150-3, /pmc/articles/PMC5691091/?report=abstract, lire en ligne)
- SEER, « Acute Myeloid Leukemia - Cancer Stat Facts », sur seer.cancer.gov (consulté le )
- Visser O , et al., « Incidence, survival and prevalence of myeloid malignancies in Europe. », sur www.ncbi.nlm.nih.gov, (consulté le )
- Global Burden of disease Study, « The global burden of childhood and adolescent cancer in 2017: an analysis of the Global Burden of Disease Study 2017 », sur www.thelancet.com, (consulté le )
- Smith MA, Ries LA, Gurney JG, et al., « Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995 », sur seer.cancer.gov, (consulté le )
- Global Burden of disease Study, « Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017 », sur www.thelancet.com, (consulté le )
- V.J. Cogliano, R. Baan, K. Straif, Y. Grosse, B. Lauby-Secretan, F. El Ghissassi, V. Bouvard, L. Benbrahim-Tallaa, N. Guha, C. Freeman, L. Galichet et C. P. Wild, « Preventable Exposures Associated With Human Cancers », JNCI Journal of the National Cancer Institute, vol. 103, no 24, , p. 1827–1839 (ISSN 0027-8874, DOI 10.1093/jnci/djr483)
- « Classification du CIRC par localisations cancéreuses | Cancer et environnement » (consulté le )
- Musée Curie, « Biographie de Marie Curie » (consulté le )
- Musée Curie, « Biographie d'Irène et Frédéric Joliot-Curie » (consulté le )
- Klervi Leuraud, David B Richardson, Elisabeth Cardis, Robert D Daniels, Michael Gillies, Jacqueline A O'Hagan, Ghassan B Hamra, Richard Haylock, Dominique Laurier, Monika Moissonnier, Mary K Schubauer-Berigan, Isabelle Thierry-Chef et Ausrele Kesminiene, « Ionising radiation and risk of death from leukaemia and lymphoma in radiation-monitored workers (INWORKS): an international cohort study », The Lancet Haematology, vol. 2, no 7, , e276–e281 (ISSN 2352-3026, DOI 10.1016/S2352-3026(15)00094-0)
- INRS, « Benzène (FT 49). Pathologie - Toxicologie - Fiche toxicologique - INRS », (consulté le )
- Loomis A., « Carcinogenicity of benzene », sur www.thelancet.com, (consulté le )
- IARC, « Benzene - IARC Monographs on the Evaluation of Carcinogenic Risks to Humans », sur iarc.fr, (consulté le )
- Kathryn Z Guyton, Dana Loomis, Yann Grosse, Fatiha El Ghissassi, Lamia Benbrahim-Tallaa, Neela Guha, Chiara Scoccianti, Heidi Mattock et Kurt Straif, « Carcinogenicity of tetrachlorvinphos, parathion, malathion, diazinon, and glyphosate », The Lancet Oncology, vol. 16, no 5, , p. 490–491 (ISSN 1470-2045, DOI 10.1016/S1470-2045(15)70134-8)
- Gabriella Andreotti, Stella Koutros, Jonathan N Hofmann, Dale P Sandler, Jay H Lubin, Charles F Lynch, Catherine C Lerro, Anneclaire J De Roos, Christine G Parks, Michael C Alavanja, Debra T Silverman et Laura E Beane Freeman, « Glyphosate Use and Cancer Incidence in the Agricultural Health Study », JNCI: Journal of the National Cancer Institute, vol. 110, no 5, , p. 509–516 (ISSN 0027-8874, DOI 10.1093/jnci/djx233)
- Centre Léon-Bérard, CIRC, « Classification du CIRC par localisations cancéreuses | Cancer et environnement », (consulté le )
- Centre National Hospitalier d'Information sur le Médicament (CNHIM), « Fiche effet indésirable », sur theriaque.org (consulté le )
- (en) Sachiko Ezoe, « Secondary Leukemia Associated with the Anti-Cancer Agent, Etoposide, a Topoisomerase II Inhibitor », International Journal of Environmental Research and Public Health, vol. 9, no 7, , p. 2444 (DOI 10.3390/ijerph9072444, /pmc/articles/PMC3407914/?report=abstract, lire en ligne)
- Barbara Deschler et Michael Lübbert, « Acute myeloid leukemia: Epidemiology and etiology », Cancer, vol. 107, no 9, , p. 2099–2107 (ISSN 0008-543X, DOI 10.1002/cncr.22233)
- LLSCanada, « www.llscanada.org » (consulté le )
- Hoffman, Ronald et al. (2005). Hematology: Basic Principles and Practice (4th. ed.). St. Louis, Mo.: Elsevier Churchill Livingstone. p. 1074–75
- Acute Myeloid Leukemia, N Engl J Med 1999;341.
- de Botton 2017, p. 500-501
- Schmidt, Cornu, Angellilo-Scherrer et al., « Bases physiopathologiques en hématologie générale : un aide-mémoire d'hématologie » [PDF], sur www.2bib.ch, (consulté le ), p. 149
- Ifrah 2018, p. 68
- Société française d'hématologie, « Leucémies aiguës, 3 - Signes biologiques et diagnostic » [html], (consulté le )
- de Botton 2017, p. 500
- (en) James W. Vardiman, « The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia », Blood, American Society of Hematology, vol. 127, no 20, , p. 2391-2405 (ISSN 1528-0020, DOI 10.1182/blood-2016-03-643544, résumé, lire en ligne)
- (en) Karen Seiter, Emmanuel C. Besa et al., « Acute Myeloid Leukemia Staging – FAB and WHO Classifications for Acute Myeloid Leukemia », sur emedicine.medscape.com, (consulté le ).
- "Update on Acute Myeloid Leukemia, septembre 2016, p 3/16"
- « Tout savoir sur le don | Don de Moelle Osseuse », sur www.dondemoelleosseuse.fr (consulté le )
- Hartmut Döhner, Elihu Estey, David Grimwade, Sergio Amadori, Frederick R. Appelbaum, Thomas Büchner, Hervé Dombret, Benjamin L. Ebert, Pierre Fenaux, Richard A. Larson, Ross L. Levine, Francesco Lo-Coco, Tomoki Naoe, Dietger Niederwieser, Gert J. Ossenkoppele, Miguel Sanz, Jorge Sierra, Martin S. Tallman, Hwei-Fang Tien, Andrew H. Wei, Bob Löwenberg et Clara D. Bloomfield, « Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel », Blood, vol. 129, no 4, , p. 424–447 (ISSN 0006-4971, DOI 10.1182/blood-2016-08-733196)
- Martin S. Tallman, Eunice S. Wang, Jessica K. Altman, Frederick R. Appelbaum, Vijaya Raj Bhatt, Dale Bixby, Steven E. Coutre, Marcos De Lima, Amir T. Fathi, Melanie Fiorella, James M. Foran, Aric C. Hall, Meagan Jacoby, Jeffrey Lancet, Thomas W. LeBlanc, Gabriel Mannis, Guido Marcucci, Michael G. Martin, Alice Mims, Margaret R. O’Donnell, Rebecca Olin, Deniz Peker, Alexander Perl, Daniel A. Pollyea, Keith Pratz, Thomas Prebet, Farhad Ravandi, Paul J. Shami, Richard M. Stone, Stephen A. Strickland, Matthew Wieduwilt, Kristina M. Gregory, Lydia Hammond et Ndiya Ogba, « Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology », Journal of the National Comprehensive Cancer Network, vol. 17, no 6, , p. 721–749 (ISSN 1540-1405, DOI 10.6004/jnccn.2019.0028)
- (en) Cancer Research UK, « Radiotherapy to the brain - Acute myeloid leukaemia », (consulté le )
- (en + es) American Cancer Society, « Radiation Therapy for Acute Myeloid Leukemia (AML) », (consulté le )
- Miguel A. Sanz, Pierre Fenaux, Martin S. Tallman, Elihu H. Estey, Bob Löwenberg, Tomoki Naoe, Eva Lengfelder, Hartmut Döhner, Alan K. Burnett, Sai-Juan Chen, Vikram Mathews, Harry Iland, Eduardo Rego, Hagop Kantarjian, Lionel Adès, Giuseppe Avvisati, Pau Montesinos, Uwe Platzbecker, Farhad Ravandi, Nigel H. Russell et Francesco Lo-Coco, « Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet », Blood, vol. 133, no 15, , p. 1630–1643 (ISSN 0006-4971, DOI 10.1182/blood-2019-01-894980)
- Gustave Roussy, « Soins de support - Gustave Roussy » (consulté le )
- Institut Curie, « Vous aider pendant les traitements Institut Curie » (consulté le )
- (en) PDQ Adult Treatment Editorial Board., « Adult Acute Myeloid Leukemia Treatment (PDQ®) - PDQ Cancer Information Summaries - NCBI Bookshelf » [html], Bethesda (MD): National Cancer Institute (US), (consulté le )
- Agence européenne des médicaments, « Dacogen », (consulté le )
- de Botton 2017, p. 504
- Agence de la biomédecine, « Activité nationale de greffe de CSH (2016) » [PDF], sur www.agence-biomedecine.fr, (consulté le )
- Carreras 2019, p. 27
- EMA, « Bulsilvex (busulfan) - Résumé des caractéristiques du produit » [PDF], (consulté le )
- Société canadienne du cancer, « Greffe de cellules souches pour la leucémie aiguë myéloblastique » (consulté le )
- Ifrah 2018, p. 77
- « Hématologie. Onco-hématologie - Présentation - EM consulte », (consulté le )
- (en) Cancer Research UK, « Total body radiotherapy (TBI) Acute myeloid leukaemia », (consulté le )
- Société de leucémie et lymphome du Canada, « Allogreffe de cellules souches » (consulté le )
- Société de leucémie et lymphome du Canada, « Greffe de cellules souches allogéniques d’intensité réduite » (consulté le )
- (en) European Society for Blood and Marrow Transplant, « Annual Report 2019 » [2019], (consulté le ), p. 68
- Jeanette Prada-Arismendy, Johanna C. Arroyave et Sarah Röthlisberger, « Molecular biomarkers in acute myeloid leukemia », Blood Reviews, vol. 31, no 1, , p. 63–76 (ISSN 0268-960X, DOI 10.1016/j.blre.2016.08.005)
- (en) American Cancer Society, « What’s New in Acute Myeloid Leukemia (AML) Research? », (consulté le )
- Elihu Estey, Judith E. Karp, Ashkan Emadi, Megan Othus et Robert Peter Gale, « Recent drug approvals for newly diagnosed acute myeloid leukemia: gifts or a Trojan horse? », Leukemia, vol. 34, no 3, , p. 671–681 (ISSN 0887-6924, DOI 10.1038/s41375-019-0704-5)
- (en) « Isocitrate dehydrogenase mutations in myeloid malignancies » (Review "Leukemia" published online 11 November 2016), Nature (revue), no 31, , p. 272–281 (DOI 10.1038/leu.2016.275, lire en ligne)
- "Ash, Clinicak News, 21 octobre 2016, AG-221 Clinical Results Promise “Revolutionary” Approach in AML"
- Starr P, « Midostaurin the First Targeted Therapy to Improve Survival in AML: Potentially Practice-Changing », Am Health Drug Benefits, vol. 9, no Spec Issue, , p. 1–21 (PMID 27014400, PMCID 4782225)
- Alexander E. Perl, Giovanni Martinelli, Jorge E. Cortes, Andreas Neubauer, Ellin Berman, Stefania Paolini, Pau Montesinos, Maria R. Baer, Richard A. Larson, Celalettin Ustun, Francesco Fabbiano, Harry P. Erba, Antonio Di Stasi, Robert Stuart, Rebecca Olin, Margaret Kasner, Fabio Ciceri, Wen-Chien Chou, Nikolai Podoltsev, Christian Recher, Hisayuki Yokoyama, Naoko Hosono, Sung-Soo Yoon, Je-Hwan Lee, Timothy Pardee, Amir T. Fathi, Chaofeng Liu, Nahla Hasabou, Xuan Liu, Erkut Bahceci et Mark J. Levis, « Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML », New England Journal of Medicine, vol. 381, no 18, , p. 1728–1740 (ISSN 0028-4793, DOI 10.1056/NEJMoa1902688)
- Arnaud Pigneux, Marie C. Béné, Philippe Guardiola, Christian Recher, Jean-Francois Hamel, Mathieu Sauvezie, Jean-Luc Harousseau, Olivier Tournilhac, Francis Witz, Christian Berthou, Martine Escoffre-Barbe, Denis Guyotat, Nathalie Fegueux, Chantal Himberlin, Mathilde Hunault, Martine Delain, Bruno Lioure, Eric Jourdan, Frederic Bauduer, Francois Dreyfus, Jean-Yves Cahn, Jean-Jacques Sotto et Norbert Ifrah, « Addition of Androgens Improves Survival in Elderly Patients With Acute Myeloid Leukemia: A GOELAMS Study », Journal of Clinical Oncology, vol. 35, no 4, , p. 387–393 (ISSN 0732-183X, DOI 10.1200/JCO.2016.67.6213)
- Agence européenne des médicaments, « VIDAZA - Résumé des caractéristiques du produit » [PDF], sur www.ema.europa.eu, (consulté le )
- Agence européenne des médicaments,, « www.ema.europa.eu » [PDF], (consulté le )
- Ceylad, « Celyad annonce une première réponse complète chez un patient atteint de LMA réfractaire et récidivante dans l’essai THINK », sur website, (consulté le )
- Yuxin Liu, Jan Philipp Bewersdorf, Maximilian Stahl et Amer M. Zeidan, « Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? », Blood Reviews, vol. 34, , p. 67–83 (ISSN 0268-960X, DOI 10.1016/j.blre.2018.12.001)
- (en) American Society of Clinical Oncology, « Leukemia - Acute Myeloid - AML: Statistics | Cancer.Net », sur www.cancer.net, (consulté le )
- (en) National Cancer Institute, « Adult Acute Myeloid Leukemia Treatment (PDQ®)–Health Professional Version », sur www.cancer.gov, (consulté le )
- (en) Cancer Research UK, « Survival | Acute myeloid leukaemia | Cancer Research UK », sur www.cancerresearchuk.org, (consulté le )
- Mareike Rasche, Martin Zimmermann, Lisa Borschel, Jean-Pierre Bourquin, Michael Dworzak, Thomas Klingebiel, Thomas Lehrnbecher, Ursula Creutzig, Jan-Henning Klusmann et Dirk Reinhardt, « Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012 », Leukemia, vol. 32, no 10, , p. 2167–2177 (ISSN 0887-6924, DOI 10.1038/s41375-018-0071-7)
- Fondation ARC, « Que sont les leucémies chez l'enfant ? | Fondation ARC pour la recherche sur le cancer », sur website, (consulté le )
- American Cancer Society, « Treatment Response Rates for Acute Myeloid Leukemia (AML) », sur www.cancer.org, (consulté le )
- Hoffman et al. 2005, p. 1071.
- Bennett JH, « Two cases of hypertrophy of the spleen and liver, in which death took place from suppuration of blood », Edinburgh Med Surg J, vol. 64, , p. 413
- (de) R Virchow, Gesammelte Abhandlungen zur Wissenschaftlichen Medizin, Francfort, Meidinger, (lire en ligne), « Die Leukämie », 190
Bibliographie modifier
![]() : document utilisé comme source pour la rédaction de cet article.
: document utilisé comme source pour la rédaction de cet article.
- Norbert Ifrah (dir. et coordonnateur), Marc Maynadié (dir. et coordonnateur) et al., Société française d'hématologie, Hématologie, Issy-les-Moulineaux, Elsevier Masson, coll. « Les Référentiels des Collèges », , 3e éd., 400 p. (ISBN 978-2-294-75108-0 et 9782294752636, EAN 9782294752636, BNF 45533912).

- R. Costello, G. Venton, J. Colle, V. Ivanov, C. Mercier, L. Delassus et V. Baccini (Article 13-018-G-50), Leucémies aiguës myéloïdes de l'adulte, Elsevier Masson, coll. « EMC / Hématologie », , 13 p. (ISBN 978-2-84299-505-8, ISSN 1155-1984, OCLC 726481102, DOI 10.1016/S1155-1984(18)83321-0, lire en ligne [html]). .
- Stéphane Vignot (dir. et rédacteur), Jean-Charles Soria (dir.) et Stéphane de Botton, Gustave Roussy & Université Paris Sud XI, Cours de chimiothérapie antitumorale et traitement médical du cancer, Paris, , 32e éd., 534 p. (ISBN 978-2-9555469-9-4), chap. 83 (« Leucémies aiguës myéloïdes »).
- (en) Enric Carreras, Carlo Dufour, Mohamad Mohty et Nicolaus Kröger, European Society for Bone Marrow Transplantation, The EBMT Handbook : Hematopoietic Stem Cell Transplantation and Cellular Therapies, Cham, Switzerland, Springer, , 688 p. (ISBN 978-3-030-02277-8 (édité erroné) et 978-3-030-02278-5, DOI 10.1007/978-3-030-02278-5, lire en ligne [PDF]).
Voir aussi modifier
Articles connexes modifier
Liens externes modifier
Grand public modifier
- Orphanet
- Guide pour les patients édité par la Société européenne d'oncologie médicale (ESMO)
- Société française d'hématologie
- cancer.be
- cancer.ca
- Fondation ARC
- Laurette Fugain, association engagée dans la lutte contre les leucémies
- (en + es) National Cancer Institute
Sites professionnels modifier
- Hématocell.fr (laboratoire d'hématologie du CHU d'Angers)
- (en) European LeukemiaNet
- (en + es) National Cancer Institute