Réaction de Grignard
Une réaction de Grignard est une réaction d'addition entre un halogénure organomagnésien et un composé organique porteur d'un groupe carbonyle, typiquement un aldéhyde ou une cétone, pour donner respectivement un alcool secondaire ou un alcool tertiaire[1]. Les halogénures organomagnésiens sont appelés réactifs de Grignard et ont pour formule générique R3–MgX, où R3 est typiquement un résidu d'alkyle, de vinyle ou d'aryle, et X représente un halogène, le plus souvent du brome Br ou du chlore Cl, parfois de l'iode I. Les réactifs de Grignard sont fortement basiques, et réduisent les aldéhydes R2–CHO et les cétones R2–CO–R1 pour former les alcools R3R2CHOH et R3R2R1COH. Une réaction de Grignard type sur une cétone R1–CO–R2 avec un bromure R3–MgBr a la forme suivante :
-
 Réaction de Grignard type.
Réaction de Grignard type.

Il s'agit d'un outil important en synthèse organique pour la formation de liaisons carbone-carbone[2],[3], mais aussi de liaisons carbone-phosphore, carbone-étain, carbone-silicium, ou encore carbone-bore. Ces réactions sont utilisées industriellement pour la production d'alcools ou encore dans l'industrie pharmaceutique. Ces composés étant sensibles aux solvants protiques, les réactions de Grignard doivent se dérouler en milieu anhydre, généralement dans l'éther diéthylique ou le tétrahydrofurane secs.
Les réactions de Grignard et les réactifs de Grignard ont été découverts en 1900 par Victor Grignard[4], chimiste français qui reçut pour cela le prix Nobel de Chimie 1912[5]. Les réactifs de Grignard sont semblables aux réactifs organolithiens car les uns comme les autres sont des nucléophiles forts capables de former de nouvelles liaisons C–C. Le caractère nucléophile est accru si l'on remplace le substituant alkyle par un groupe amidure. De tels amidures d'halogénures de magnésium sont appelés bases de Hauser (en).
Réactions de Grignard et applications modifier
L'électronégativité du carbone, qui vaut χC = 2,55 sur l'échelle de Pauling, est supérieure à celle des substituants métalliques, de sorte que la liaison carbone–métal est polarisée –Cδ–Mδ+ ; cet atome carbone se comporte alors en nucléophile, attaquant l'atome de carbone électrophile présent dans une liaison polaire d'un groupe carbonyle. On pense que la réaction d'addition correspondante fait généralement intervenir deux molécules de réactif de Grignard formant un intermédiaire réactionnel cyclique à six atomes[6].
-
 Mécanisme d'une réaction de Grignard.
Mécanisme d'une réaction de Grignard.

Les réactifs de Grignard encombrés peuvent agir par transfert mono-électronique. On suppose qu'il existe des voies de synthèse semblables pour les autres réactions induites par ces réactifs de Grignard, comme la formation de liaisons carbone-phosphore, carbone-étain, carbone-silicium, ou encore carbone-bore.
Les réactions de Grignard se déroulent généralement en solution dans un éther, particulièrement l'éther éthylique et le tétrahydrofurane.
Une limitation des réactifs de Grignard est qu'ils ne réagissent pas facilement avec les halogénures d'alkyle via un mécanisme de substitution nucléophile bimoléculaire (SN2). Ils participent en revanche facilement aux réactions de transmétallation :
- R–MgX + AlX → Al–R + MgX2.
Les réactifs de Grignard disponibles dans le commerce sont particulièrement utiles dans cet usage car cette voie de synthèse résout le problème de l'amorçage de la réaction[7].
Il est important d'éliminer toute trace d'eau et d'air du milieu dans lequel on conduit une réaction de Grignard, car les réactifs de Grignard sont rapidement détruits par protonolyse (en) ou par oxydation[8]. Dans la mesure où la plupart de ces réactions sont menées dans l'éther diéthylique ou le tétrahydrofurane anhydres, les réactions parasites dues à l'air sont limitées par la couche protectrice formée par les vapeurs du solvant. Il est recommandé de mener ces réactions sous atmosphère inerte d'azote ou d'argon.
Les réactions de Grignard sont susceptibles de donner des alcools primaires, secondaires ou tertiaires, selon le type de carbonyle employé. Les esters donnent des alcools tertiaires par addition de deux équivalents de réactifs de Grignard en formant une cétone intermédiaire, laquelle réagit immédiatement avant de pouvoir être isolée. Avec le dioxyde de carbone CO2 il se forme un acide carboxylique R3COOH. Outre les aldéhydes, les cétones et les esters, il est également possible d'utiliser des réactifs de Grignard avec les nitriles –C≡N, les imines >C=N–, les époxydes, les thioesters, etc.
-
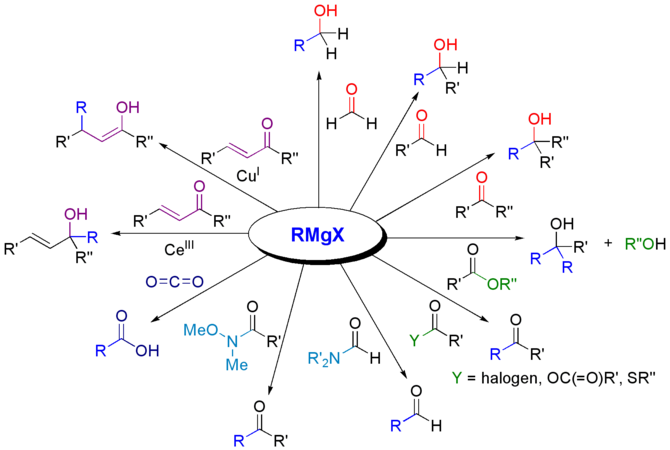 Réactions de Grignard avec des composés carbonylés.
Réactions de Grignard avec des composés carbonylés. -
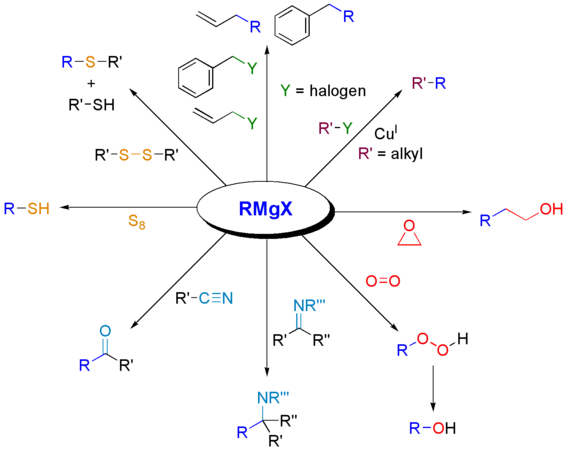 Réactions de Grignard avec divers autres électrophiles.
Réactions de Grignard avec divers autres électrophiles.


La réaction de Grignard la plus couramment mise en œuvre est l'alkylation des aldéhydes et des cétones, par exemple à l'aide de bromure de vinylmagnésium[9] CH2=CH–MgBr :
-
 Exemple d'alkylation d'un composé organique par le bromure de vinylmagnésium.
Exemple d'alkylation d'un composé organique par le bromure de vinylmagnésium.

Les réactions de Grignard trouvent des applications industrielles multiples. Jusqu'à l'interdiction de ces additifs pour carburants, elles ont joué un rôle important dans la production de composés de type tétraéthylplomb[10] Pb(C2H5)4. Aujourd'hui, la synthèse de plusieurs médicaments fait intervenir des réactifs de Grignard, comme la production du naproxène ou celle du tamoxifène :
-
 Étape finale de la synthèse du naproxène, par une réaction de Grignard.
Étape finale de la synthèse du naproxène, par une réaction de Grignard.

-
![Étape de la synthèse du tamoxifène impliquant le bromure de phénylmagnésium, un réactif de Grignard[11].](//upload.wikimedia.org/wikipedia/commons/thumb/9/94/TamoxifenSynthesisGrignard.svg/561px-TamoxifenSynthesisGrignard.svg.png) Étape de la synthèse du tamoxifène impliquant le bromure de phénylmagnésium, un réactif de Grignard[11].
Étape de la synthèse du tamoxifène impliquant le bromure de phénylmagnésium, un réactif de Grignard[11].
![Étape de la synthèse du tamoxifène impliquant le bromure de phénylmagnésium, un réactif de Grignard[11].](/wiki/Fichier:TamoxifenSynthesisGrignard.svg)
Outre les liaisons carbone-carbone, les réactions de Grignard permettent de produire des liaisons entre le carbone et d'autres éléments. Les composés organoborés utilisés dans les réactions de Suzuki sont à cet égard particulièrement notables[12].
-
 Réactions de Grignard formant des liaisons entre le carbone et divers hétéro-atomes.
Réactions de Grignard formant des liaisons entre le carbone et divers hétéro-atomes.

Les réactions avec les halogénures métalliques permettent également de produire des liaisons entre le carbone et des hétéro-atomes. Les réactifs de Grignard réagissent en effet avec de nombreux électrophiles métalliques, comme l'illustre la transmétallation avec le chlorure de cadmium CdCl2 donnant un dialkylcadmium :
- 2 R1MgX + CdCl2 → R12Cd + 2 MgXCl.
Les composés dialkylcadmium sont utilisés pour la préparation de cétones à partir d'halogénures d'acyles :
- 2 R2C(O)Cl + R12Cd → 2 R2C(O)R1 + CdCl2.
Contrairement à ce qu'on observe avec les halogénures métalliques, les réactifs de Grignard ne réagissent généralement pas avec les halogénures organiques. Ils peuvent en revanche participer à des réactions de couplage C–C en présence de catalyseurs métalliques. Ainsi, le bromure de nonylmagnésium CH3(CH2)8MgBr réagit avec le para-chlorobenzoate de méthyle ClC6H4COOCH3 pour donner l'acide para-nonylbenzoïque CH3(CH2)8C6H4COOH en présence de tris-acétylacétonate de fer(III) Fe(acac)3, après traitement à l'hydroxyde de sodium NaOH pour hydrolyser l'ester ; en l'absence du catalyseur ferrique, le réactif de Grignard attaquerait le groupe ester sur l'halogénure d'acyle[13].
-
 Synthèse de l'acide para-nonylbenzoïque par une réaction de Grignard.
Synthèse de l'acide para-nonylbenzoïque par une réaction de Grignard.

Le chlorure de nickel NiCl2 dans le tétrahydrofurane est également un bon catalyseur pour le couplage des halogénures d'aryle avec des réactifs de Grignard. Le tétrachlorocuprate de dilithium Li2CuCl4 est également un catalyseur efficace pour réaliser le couplage d'halogénures d'alkyle : on l'obtient en mélangeant du chlorure de lithium LiCl et du chlorure de cuivre(II) CuCl2 dans le THF. Le couplage de Kumada permet d'obtenir des styrènes substitués.
Notes et références modifier
- (en) Michael B. Smith, Jerry March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6e édition, Wiley-Interscience, New-York, 2007. (ISBN 0-471-72091-7)
- (en) David A. Shirley, « The Synthesis of Ketones from Acid Halides and Organometallic Compounds of Magnesium, Zinc, and Cadmium », Organic Reactions, , p. 28-58 (DOI 10.1002/0471264180.or008.02, lire en ligne)
- (en) Donna M.Huryn, « 1.2 - Carbanions of Alkali and Alkaline Earth Cations: (ii) Selectivity of Carbonyl Addition Reactions », Comprehensive Organic Synthesis, vol. 1, , p. 49-75 (DOI 10.1016/B978-0-08-052349-1.00002-0, lire en ligne)
- Victor Grignard, « Sur quelques nouvelles combinaisons organométalliques du magnésium et leur application à des synthèses d'alcools et d'hydrocarbures », Comptes rendus hebdomadaires des séances de l'Académie des sciences, vol. 130, , p. 1322-1325 (lire en ligne)
- (en) « The Nobel Prize in Chemistry 1912 » (consulté le ).
- (en) Kazuhiro Maruyama et Toshimasa Katagiri, « Mechanism of the Grignard reaction », Journal of Physical Organic Chemistry, vol. 2, no 3, , p. 205-213 (DOI 10.1002/poc.610020303, lire en ligne)
- (en) G. S. Silverman et P. E. Rakita, Handbook of Grignard Reagents, CRC Press, 1996. (ISBN 0824795458)
- (en) M. T. Goebel et C. S. Marvel, « The Oxidation of Grignard Reagents », Journal of the American Chemical Society, vol. 55, no 4, , p. 1693-1696 (DOI 10.1021/ja01331a065, lire en ligne)
- (en) Jarle André Haugan, « Total Synthesis of C31-Methyl Ketone Apocarotenoids 2: The First Total Synthesis of (3R)-Triophaxanthin », Acta Chemica Scandinavica, vol. 51, , p. 1096-1103 (DOI 10.3891/acta.chem.scand.51-1096, lire en ligne)
- (en) C. Elschenbroich, A. Salzer, Organometallics – A Concise Introduction, 2e édition, Wiley-VCH, Weinheim, 1995, p. 139. (ISBN 3-527-28164-9).
- (en) Herman Glenn Richey, Grignard Reagents: New Developments, Wiley, 2000. (ISBN 0471999083)
- (en) A. Suzuki, Metal-Catalyzed Cross-coupling Reactions, édité par P. J. Stang et F. Diedrich, Wiley-VCH, Weinheim, 1998.
- (en) Alois Fürstner, Andreas Leitner et Günter Seidel, « 4-nonylbenzoic acid », Organic Syntheses, vol. 81, , p. 33 (DOI 10.15227/orgsyn.081.0033, lire en ligne)