Atropoisomère
Un atropoisomère ou atropisomère est un cas spécial d'énantiomère axial dû au blocage de la rotation autour d'une liaison simple (rotamérie). Il s'agit d'une sous-classe de rotamères dans laquelle la barrière énergétique due à l'encombrement stérique est tellement élevée qu'elle permet d'isoler les rotamères[1]. Le terme atropoisomère est dérivé de l'« a » grec privatif et de « tropos » qui signifie tourner, accolé à isomère. Ce nom, atropoisomère, a été inventé par R. Kuhn en 1933[2] bien que l'atropoisomérie ait été détectée pour la première fois sur l'acide 6,6'-dinitro-2,2'-diphénique par Christie en 1922[3]. M. Oki définit comme atropoisomère, des conformères qui s'interconvertissent avec une demi-vie de plus de 1000 secondes (16 min 40 s) à une température donnée[4].

Les atropoisomères constituent une classe importante de composés chimiques de chiralité axiale. Ils se distinguent des autres composés chiraux en ce qu'ils peuvent être équilibrés (racémisés) thermiquement comme les invertomères tandis que pour les autres types d'énantiomères ce n'est généralement possible que par réarrangement chimique.
La classe la plus importante d'atropoisomère est celle des biaryles tels que les acides diphéniques substitués, des dérivés du biphényle avec des substituants sur l'ensemble de ses positions ortho. D'autres sont des dimères de dérivés de naphtalène tels que le 1,1'-bi-2-naphtol ou BINAP qui est un ligand pouvant être utilisé pour la préparation de stéréoisomères optiquement actifs. De la même manière, des systèmes aliphatiques cycliques comme des cyclohexanes liés par une liaison simple peuvent présenter une atropoisomérie s'ils ont des substituants assez volumineux pour bloquer leur orientation relative.
La séparation d'atropisomères peut être faite par les méthodes de résolution chirale courantes telles que la cristallisation sélective.
Dans une synthèse atropo-énantiosélective ou atroposélective, un atropoisomère est privilégié par rapport à l'autre. Les synthèses atroposélectives peuvent être réalisées en utilisant des auxiliaires chiraux, comme le catalyseur CBS dans la synthèse totale de la knipholone, ou en utilisant des approches basées sur l'équilibration thermodynamique quand une réaction d'isomérisation favorise un atropoisomère sur l'autre.
Des exemples d'atropoisomères naturels sont la vancomycine ou la knipholone, qui se trouve dans les racines de Kniphofia foliosa de la famille des Asphodelaceae.
-
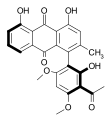 knipholone
knipholone -

-
 télenzépine
télenzépine



Un autre exemple d'atropisomérie est le pesticide métolachlore qui présente le blocage de la rotation autour d'une liaison C-N en plus d'un atome de carbone asymétrique. Seuls les diastéréoisomères (S,Sa) et (S,Ra) ont des propriétés herbicides[5],[6]. La télenzépine est aussi atropoisomérique, car la molécule a également une liaison C-N stéréogène. En solution aqueuse neutre, elle affiche une demi-vie pour la racémisation de l'ordre de 1000 ans. Les énantiomères ont été résolus. L'isomère dextrogyre est environ 500 fois plus actif que l'isomère lévogyre au niveau des récepteurs muscariniques dans le cortex cérébral du rat[7].
Exemple d'application modifier
L'asymétrie d'un atropoisomère est transférée dans une réaction chimique à un nouveau stéréocentre[8]. L'atropoisomère est un composé iodoaryl synthétisé à partir de la (S)-valine et existe sous la forme des isomères (M,S) et (P,S) du fait du blocage de la rotation autour de la liaison CN. La barrière d'interconversion entre les deux est de 101,7 kJ/mol (24,3 kcal/mol). L'isomère (M,S) peut être obtenu pur à partir du mélange par recristallisation dans un mélange d'hexanes. Afin de former un radical aryle, l'atome d'iode est éliminée homolytiquement grâce à un mélange d'hydrure de tributylétain, de triéthylbore et d'oxygène comme dans une réaction de désoxygénation de Barton-McCombie (en). Bien que la rotation empêchée soit supprimée dans le radical aryle, la réaction intramoléculaire avec l'alcène est suffisamment plus rapide que la rotation autour de la liaison CN pour que la stéréochimie soit préservée. De cette façon, l'isomère (M,S) forme exclusivement l'énantiomère (S,S)-dihydroindolone.

transfert d'asymétrie par l'intermédiaire d'atropoisomères
Un commutateur de chiralité axiale a été rapporté pour un diol préparé à partir d'un couplage pinacolique intramoléculaire du dérivé di-aldéhyde correspondant avec l'iodure de samarium(II)[9]. Dans le méthanol, ce composé possède deux fonctions alcool en position équatoriale mais dans l'hexane, l'hélicité est inversée et les deux groupes sont en position axiale.

Notes et références modifier
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Atropisomer » (voir la liste des auteurs).
- (en) Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M, « Atroposelective synthesis of axially chiral biaryl compounds », Angewandte Chemie International Edition, vol. 44, no 34, , p. 5384–5427 (PMID 16116589, DOI 10.1002/anie.200462661).
- (en) Kuhn R, Stereochemie, Frendenberg, K. Ed.; Franz Deutike, , « Molekulare asymmetrie », p. 803.
- Christian Wolf, Dynamic stereochemistry of chiral compounds: principles and applications, The Royal Society of Chemistry, Cambridge 2008, p. 84.
- M. Oki, Topics in Stereochemistry, 1983, 1.
- H.-U. Blaser, The chiral switch of (S)-metolachlor: A personal account of an industrial odyssey in Asymmetric Catalysis, Advanced Synthesis and Catalysis, 2002, vol. 344, pp. 17–31.
- Hans-Ulrich Blaser, Industrielle asymmetrische Hydrierung „Made in Switzerland“, Nachrichten aus der Chemie, 2010, vol. 58, pp. 864−867.
- J. Clayden, W. J. Moran, P. J. Edwards, S. R. LaPante, The challenge of atropisomerism in drug discovery, Angewandte Chemie International Edition, 2009, vol. 48(35), pp. 6398-6401. DOI 10.1002/anie.200901719, .
- Marc Petit, Andre J. B. Lapierre, and Dennis P. Curran, Relaying asymmetry of transient atropisomers of o-iodoanilides by radical cyclizations, J. Am. Chem. Soc., 2005, vol. 127(43), pp. 14994-14995, (Communication). DOI 10.1021/ja055666d, Abstract.
- Stefan Reichert and Bernhard Breit, Development of an axial chirality switch, Org. Lett., 2007, vol. 9(5), pp. 899-902, (Letter). DOI 10.1021/ol0700660.